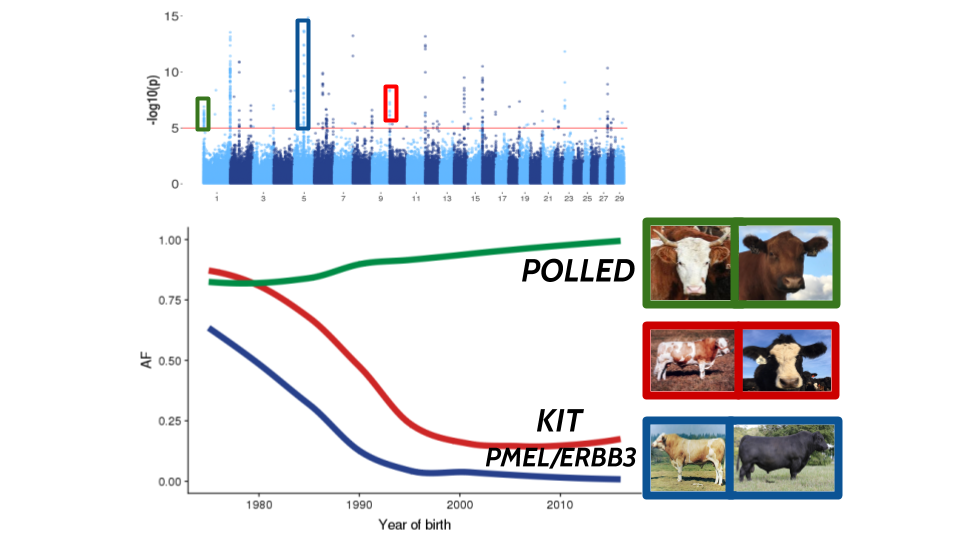
I am interested in understanding how artificial and natural selection on complex traits change the genome over time. Most methods for detecting selection rely on identifying allele frequency differences between diverged populations (like FST or FLK) or on genomic signatures that have developed due to rapid allele frequency changes (like measures of haplotype homozygosity or singleton density). These methods have had success in identifying selective sweeps on loci affecting simple traits (e.g. POLLED, MC1R, KIT) or very large-effect QTL (e.g. DGAT, LCORL, PLAG1). These methods struggle to detect ongoing polygenic selection within homogeneous populations (like cattle in the US).
We’ve developed a simple method, Generation Proxy Selection Mapping (GPSM), for detecting polygenic selection within a population that has multiple generations of genotype data. We assume that alleles under selection will be at a significantly different frequency in recent compared to distant generations. GPSM is essentially a GWAS for generation number. Instead of a continuous measured phenotype, we fit some proxy for generation number (birth year, selection cycle, radiocarbon date, etc.) as the dependent variable. Using a linear mixed model allows us to control for potential confounding population and family structure.
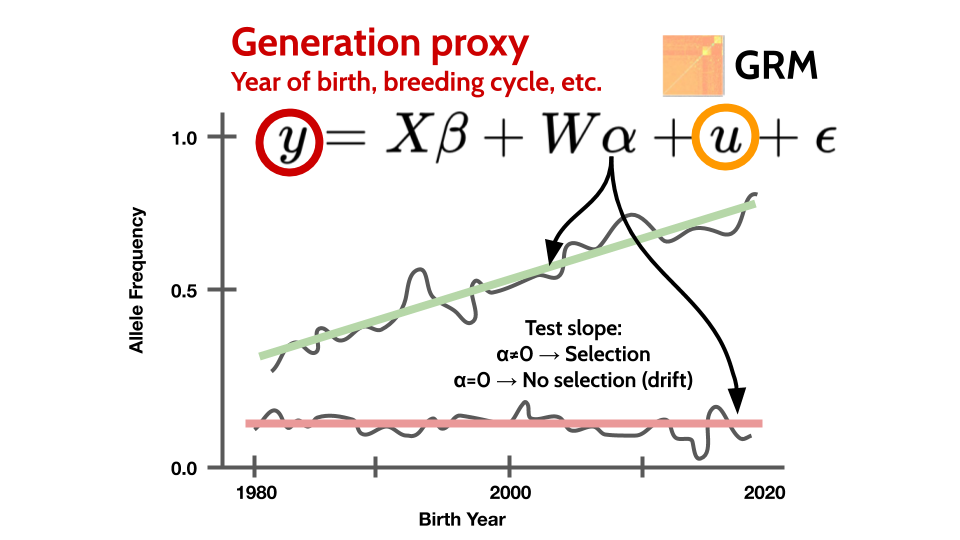
GPSM tests for changes in allele frequency greater than would be expected due to drift. A linear mixed model controls for uneven sampling of generations and family structure.
When applied to U.S. beef cattle populations we identify dozens of genomic regions under selection. These regions overlap known Mendelian loci, large-effect QTL, and novel targets of selection. Our real data and simulations both suggest that GPSM is able to identify exceedingly small allele frequency changes over very short periods of time, consistent with ongoing polygenic selection.